
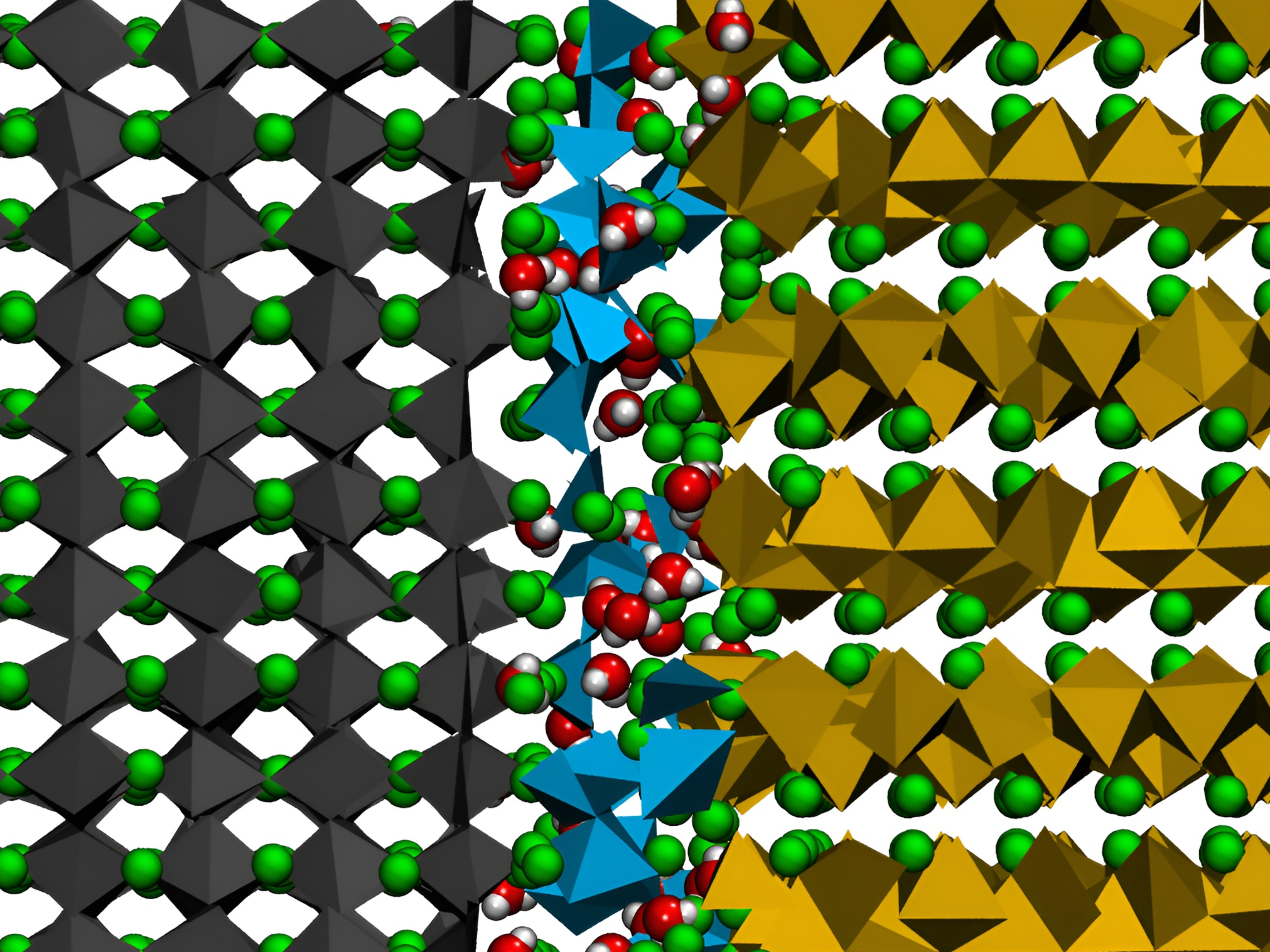
探究晶体结构转变
当碳原子以六角排列时,便形成了柔软乌黑的石墨;而当它们以四面体排列时,则成为了璀璨坚硬的钻石。晶体的性质随着晶体结构的转变发生着显著的变化。我们着迷于探索发生在晶体材料中的各种结构转变。利用密度泛函理论(DFT)、量化(QM)计算以及分子动力学(MD)模拟等方法,我们探究晶体结构转变的机理以及结构转变所带来的材料性质变化。我们目前聚焦于无机钙钛矿晶体中的多种固-固结构转变。理解这类重要材料的晶体结构转变的热力学平衡和动力学过程,可为提升其稳定性提供重要的理论指导。
Exploring Crystal Structure Transformations
The properties of crystals undergo significant changes as their structures transform. We are captivated by exploring various structural transformations occurring in crystalline materials. Employing molecular simulation techniques such as Density Functional Theory (DFT), Quantum Mechanical (QM) calculations, and Molecular Dynamics (MD) simulations, we delve into the mechanisms behind these transformations and the correlating changes in material properties. Our current focus centers on diverse solid-solid structural transformations in inorganic perovskite crystals. Understanding the thermodynamic equilibrium and kinetic processes of these transformations provides important theoretical guidance for enhancing the stability of these important materials.
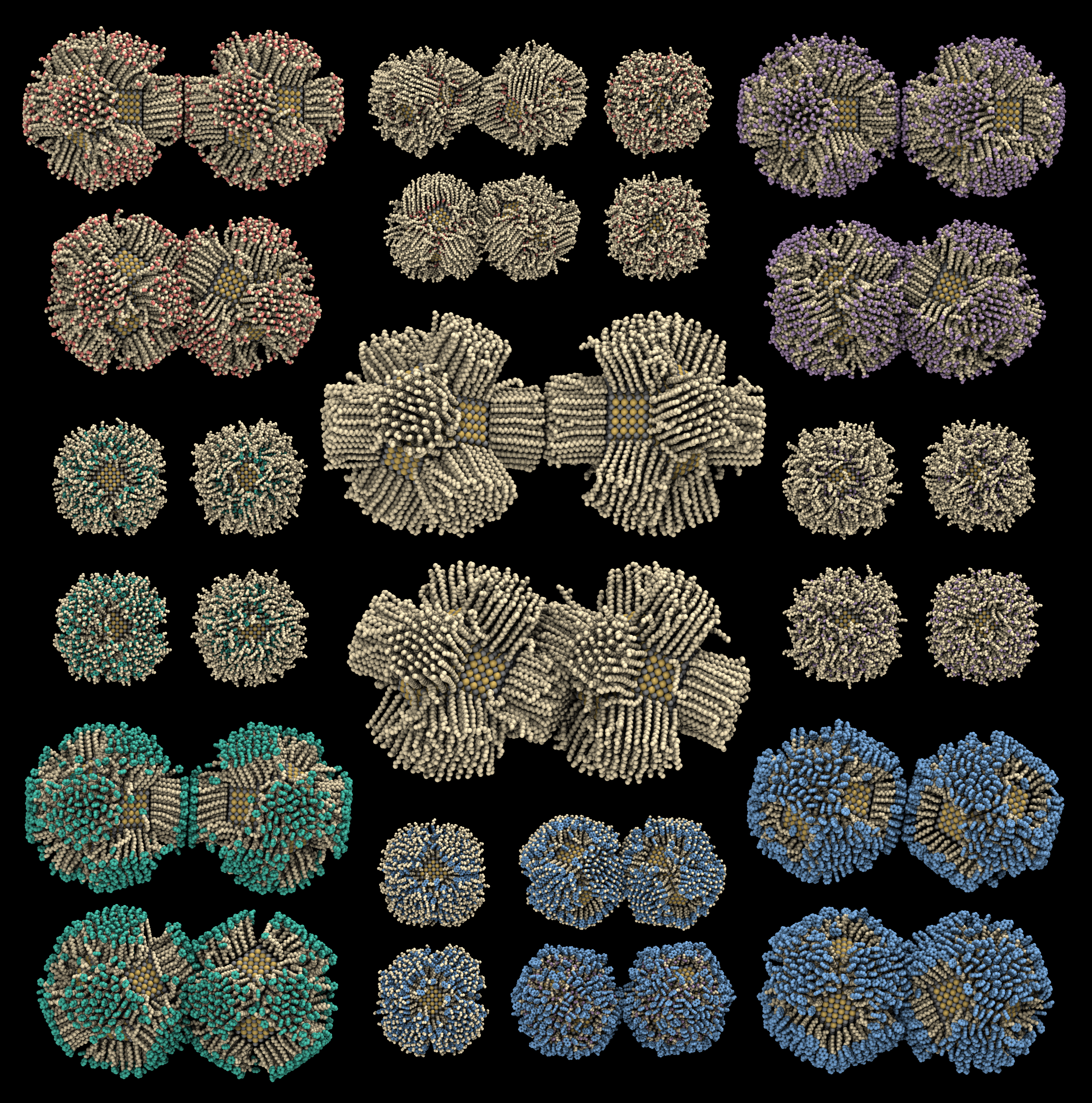
胶体纳米颗粒的表面化学与可控组装
无机纳米颗粒通过表面附着有机配体分子,得以溶解形成胶体溶液。通过控温、调节溶剂等方法,可以使分散的纳米颗粒慢慢析出并排列组装成长程有序的超晶体。这种以纳米颗粒为“人工原子”制备“人工晶体”的方法,已逐渐成为自下而上设计先进材料的新范式。我们利用分子模拟结合增强采样及粗粒化方法,探究胶体纳米颗粒表面化学对颗粒间相互作用及颗粒动力学的影响。通过计算机模拟,设计可控组装的胶体纳米颗粒,创造具有新颖结构和特殊性质的“人工晶体”新材料。
Surface Chemistry and Controlled Assembly of Colloidal Nanoparticles
Inorganic nanoparticles capped with organic ligands on their surfaces dissolve to form colloidal solutions. By manipulating temperature and solvent conditions, dispersed nanoparticles slowly precipitate and assemble into long-range ordered supercrystals. This method, using nanoparticles as "artificial atoms" to create "artificial crystals," has emerged as a novel paradigm for the bottom-up design of advanced materials. Utilizing molecular simulations, together with enhanced sampling and coarse-graining methods, we explore the relationship between nanoparticle surface chemistry, inter-particle interactions, and dynamics behaviors. We design controllable assemblies of colloidal nanoparticles through computer simulations, creating "artificial crystals" with novel structures and unique properties.
机器学习与粗粒化方法
传统DFT计算仅适用于数十个原子的体系,而几万原子的MD模拟又常因模型不准确而难以得到正确结果。分子模拟中计算精度与计算速度长期以来是一对难以调和的矛盾体。然而,机器学习和粗粒化方法的快速发展为解决这一问题提供了可能性,有望实现对大体系、长时间动力学的精确模拟。我们致力于通过训练神经网络准确描述粒子间多体相互作用,开发准确的隐式溶剂模型,并应用神经网络模型于隐世溶剂模型,以实现溶液中纳米构筑单元动力学行为的准确预测,实现从原子到介观尺度的精密链接。
Machine Learning and Coarse-Graining Methods in Dynamics Simulation
Traditional DFT calculations are limited to systems of a few tens of atoms, while MD simulations involving tens of thousands of atoms may suffer inaccuracies due to model limitations. The trade-off between computational accuracy and speed has long been a challenging dichotomy in molecular simulations. However, the rapid development of machine learning and coarse-graining methods offers a potential resolution, enabling precise simulations of large systems and long-time dynamics. Our commitment involves training neural networks to accurately describe inter-particle interactions, developing precise implicit solvent models, and applying neural network models to implicit solvent models. This approach aims to achieve accurate predictions of the dynamics of nanoscale building blocks, establishing a precise link from the atomic to mesoscopic scale.

课题组长
樊炤川 研究员
邮件:zcfan2021@sinano.ac.cn
电话:(0512) 62872936
办公地址:D815
教育与科研经历
博士,代尔夫特理工大学,代尔夫特,荷兰(2011-2016)
博士后,犹他大学,盐湖城,美国(2015-2019)
博士后,科廷大学,珀斯,澳大利亚(2019-2020)
研究员,中国科学院苏州纳米所,苏州(2021-至今)
Principal Investigator
Dr. Zhaochuan Fan
Research Professor
Email: zcfan2021@sinano.ac.cn
Phone: +86 (512) 6287-2936
Office: D815
Education & Research Experience
- Ph.D., Delft University of Technology, Delft, Netherlands (2011–2016)
- Postdoc, University of Utah, Salt Lake City, USA (2015–2019)
- Postdoc, Curtin University, Perth, Australia (2019–2020)
- Research Professor, SINANO, CAS, Suzhou, China (2021–Present)
Publications: See Google Scholar or ORCID profile.
